Explanations
a. Downgraded by two levels for risk of bias: Dmitri Puskar, Holubar M, Solayamani Dodaran M, Shenoy S, Udwadia Z and Zhao H have some concerns and Balykova, Behnam Mahmudi, Chen, Finberg, Lou, Ruzhenstova TA, Shinkai M and Tabarsi have high RoB
b. Downgraded by 1 level for inconsistency: As we identified moderate heterogeneity, (I² = 59%)
c. Downgraded by one level for serious imprecision: lower confidence interval bound represents a mild benefit from Favipiravir whereas the upper bound includes harm.
d. Downgraded by one level for Risk of Bias as Ivaschenko and Solayamani Dodaran M had some concerns in the risk of bias whereas Behnam Maahmudie, Finberg, Lou Y and Ruzhenstova (40%) have high risk of bias.
e. Not downgraded for inconsistency as we did not identify any heterogeneity( I²=0%)
f. Downgraded by 2 levels for Risk of Bias, as Dmitry Pushkar has some concerns and Finberg has high Risk of Bias.
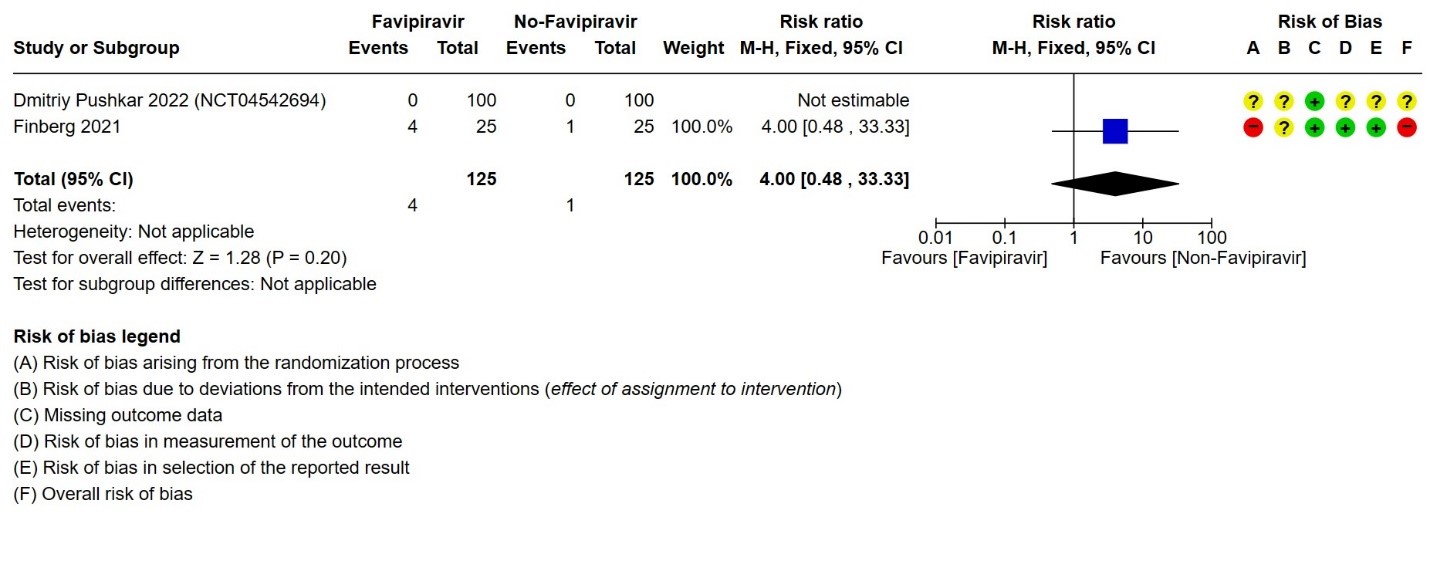
g. Downgraded by 2 levels for very serious Imprecision as the lower bound represents appreciable benefit and upper bound suggests harm (0.48, 33.33)
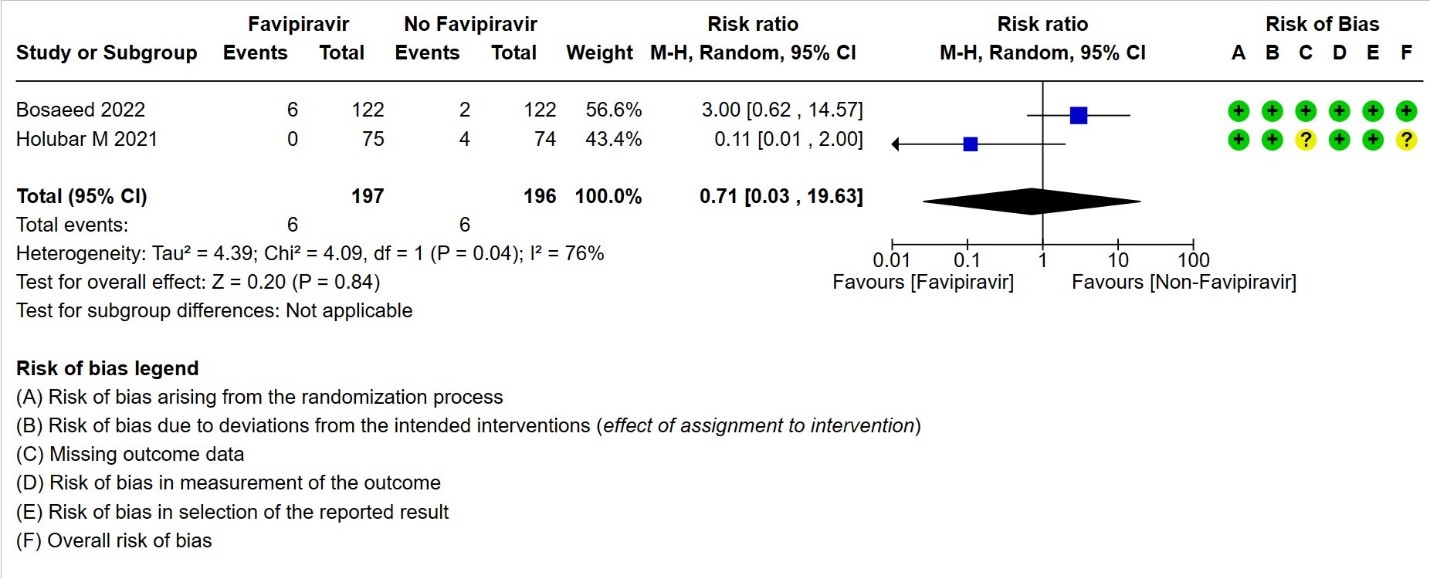
h. Downgraded two levels due to inconsistency. We identified considerable heterogeneity (I² = 76%).
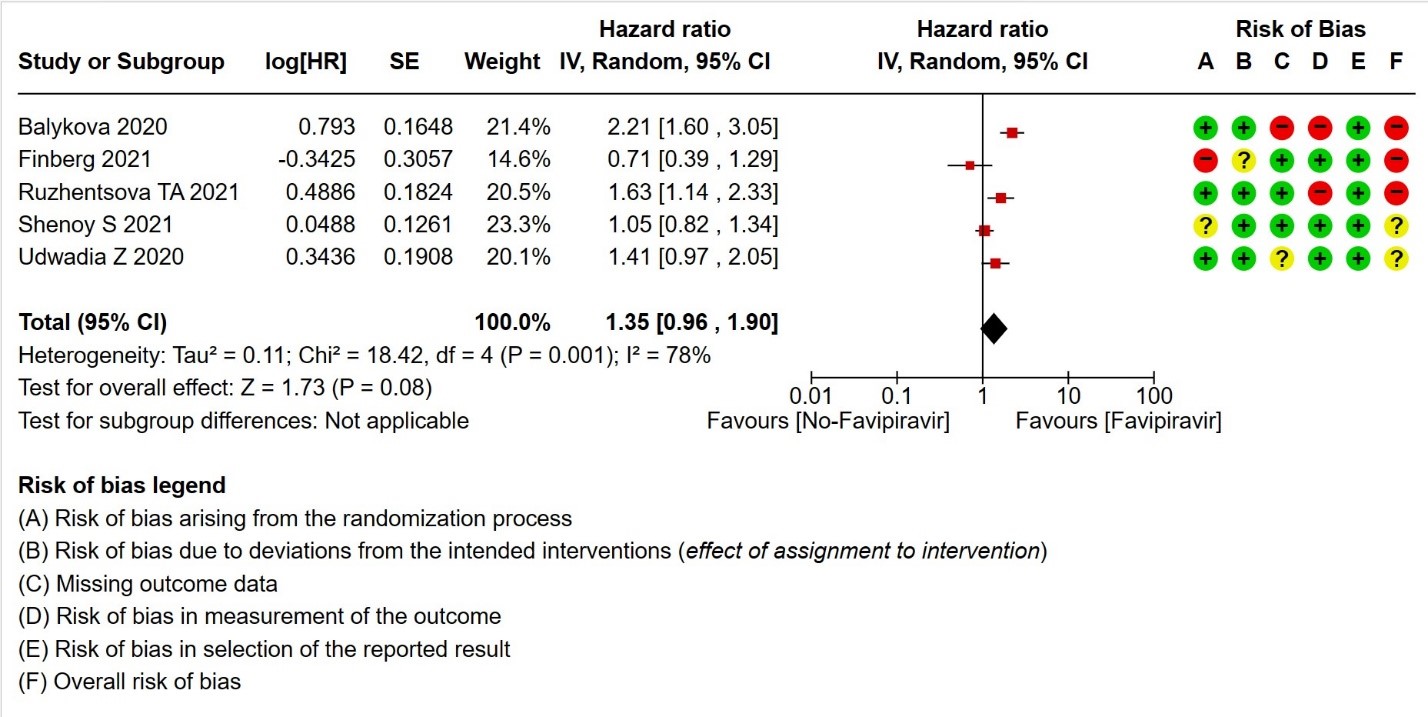
i. Downgraded by two levels for very serious risk of bias: Balykova reported high risk of bias with regard to missing outcomes and measurement of outcomes; Finberg reported high risk of bias arising from the randomization process and some concerns due to deviations from intended interventions; Ruzhenstova reported high risk of bias due to measurement of outcome; Shenoy reported some concerns due to bias arising from randomization process; Udwadia reported some concerns due to missing outcome data.
j. Downgraded two levels due to inconsistency. We identified considerable heterogeneity (I² = 78%).
k. Downgraded by one level for serious imprecision: lower confidence interval bound represents mild harm from Favipiravir, whereas the upper bound includes appreciable benefit.
l. Downgraded by one level for serious risk of bias: Lou reported a high risk of bias in randomization and some concerns due to deviations from intended interventions.
m. Downgraded by two levels for very serious risk of bias: Ruzhenstova reported a high risk of bias due to measurement of outcome; Solaymani reported some concerns due to bias arising from the selection of reported results whereas Tabarsi reported a high risk of bias arising from deviation from intended interventions, missing outcome data and some concerns due to selection of reported results.
n. Downgraded by one level for serious imprecision: lower confidence interval bound represents an appreciable benefit from Favipiravir whereas the upper bound includes harm.
o. Not downgraded for risk of bias as Chuah H has no risk of bias (92.2% weightage) whereas as studies with minimal weightage like Behnam Mahmudie and Finberg has high risk of bias
p. Not downgraded for inconsistency as we did not identify any important heterogeneity(I²=14%)
q. Downgraded by one level for imprecision: the optimal information size is not met
r. Downgraded by 2 levels for Risk of Bias as Shinkai, Ruzhentsova and Finberg (>40%) have high risk of bias.
s. Downgraded by 1 level for inconsistency: As we identified moderate heterogeneity, (I² = 52%)
t. Downgraded by one level for serious imprecision: lower confidence interval bound represents mild harm from Favipiravir, whereas the upper bound suggests appreciable benefit.
u. Downgraded by 2 levels for Risk of Bias as Balykova, Ruzhnetsova, Shinkai, Chen & Finberg contribute to >50% of weightage and has high risk of bias
v. Downgraded by 1 level for inconsistency: As we identified considerable heterogeneity, (I² = 72%)
w. Not downgraded for Imprecision, even though the confidence of interval varies from 1.25 to 1.61 because the upper and lower bound point towards harm from the intervention.
x. Downgraded by one level for risk of bias: Holubar M, Shenoy S, Udwadia Z and Zhao H has some concerns whereas Balykov, Finberg, Lou Y, Ruzhenstova TA and Shinkai S have high risk of bias
y. Not downgraded for inconsistency as we did not identify any important heterogeneity(I²=46%)
The World Health Organization (WHO) on March 11, 2020, declared COVID-19 a worldwide pandemic because of its widespread prevalence and life-altering consequences (1). The disease is caused by SARS-CoV-2 Virus that largely depends on angiotensin-converting enzyme 2 (ACE-2) receptor binding, RNA-dependent RNA polymerase (RdRp), as well as other host and viral proteins important for successful transmission and replication (2). Several interventions have emerged in the past two years for treating COVID-19, along with numerous existing antivirals currently being investigated in terms of their suitability as potential treatment options, a simple oral antiviral drug for COVID-19 has not yet been developed. (3–5)
Favipiravir (FVP) is one of the many antivirals that are currently being investigated for their efficacy and safety in Covid-19. It is a broad-spectrum antiviral discovered in Japan and was initially used in the management of influenza, Ebola, and other viral diseases. FVP is a nucleoside analog that can be triphosphorylated in cells to become active and serves as a substrate of virus RNA-dependent RNA polymerase (RdRp), leading to an increased mutation frequency, and possibly lethal mutagenesis (6). An excellent bioavailability of ~94%, 54% protein binding, and short half-life with rapid elimination makes favipiravir a promising candidate in the treatment of covid 19. (7)
There is conflicting evidence on the efficacy and safety of favipiravir in Covid 19. FVP was shown to inhibit SARS CoV-2 in vitro (5,8), however, the clinical benefits in Covid-19 patients are relatively low and not established (9,10).With the availability of more studies and the inconsistencies in the data and it is imperative to conduct an updated systematic review to evaluate the clinical benefits of FVP as a treatment for Covid-19
This guideline recommendation was made according to the evidence synthesized by the authors for the systematic review conducted according to the Cochrane protocol available in https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD015219/full
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Epistemonikos, and the COVID‐19‐specific resource www.covid‐nma.com, for studies of any publication status and in any language published up to September 2022. We also reviewed reference lists of systematic reviews and included studies. We contacted researchers to identify unpublished and ongoing studies.
We included randomized controlled trials (RCTs) testing Favipiravir treatment of any dose or duration in people with acute COVID‐19, whether suspected or confirmed. Trials were included if the intervention arm had Favipiravir with or without another experimental drug, and if the comparator arm did not include Favipiravir (this could involve use of placebo, standard care, or other potentially active drugs). We excluded trials that did not report any outcomes that could provide usable data for the review, those which were quasi-randomized and those lacking a comparator arm.
We extracted data for the following outcomes, pre-defined by the Expert Working Group:
- Critical (primary for this review):
- Progression to:
- Oxygen therapy
- Ventilation: non-invasive or invasive
- Critical or Intensive care (any reason)
- Duration of hospitalization
- Progression to:
- Important (secondary):
- Mortality (all-cause) – at 28-30 days, or in-hospital
- Time for clinical improvement
- Time to negative PCR for SARS-CoV-2
- Complications of COVID-19:
- Thrombotic events
- Pulmonary function/fibrosis
- Long covid/post-acute sequelae
- Secondary infections
- Adverse events:
- All and serious, hyperuricemia
Two reviewers independently assessed the eligibility of search results using the online Rayyan tool. Data extraction was performed by one reviewer, and checked by another, using a piloted data extraction tool on Microsoft Word. Risk of bias (RoB) was assessed using the Cochrane RoB v2.0 tool, with review by a second reviewer and compared with covid-nma database. If there was a difference in more than one domain, it was assessed by a third independent author. We planned to use risk ratios (RR) for dichotomous outcomes and mean differences (MD) for continuous outcomes, with 95% confidence intervals (CIs). We planned to meta-analyze if appropriate, i.e., if outcomes were measured and reported in a similar way for similar populations in each trial, using random-effects models with the assumption of substantial clinical heterogeneity (the appropriateness of which would be checked by the Methodology Committee), and the I2 test to calculate residual statistical heterogeneity, using Review Manager (RevMan) v5.4. The sensitivity analysis was done by comparing after splitting into 2 categories -SOC + active comparator (another antiviral) vs together. We performed sensitivity analysis for outcomes with high I2 values by incorporating only trials with Low RoB. We used GRADE methodology to make a summary of findings tables on GradeProGDT.
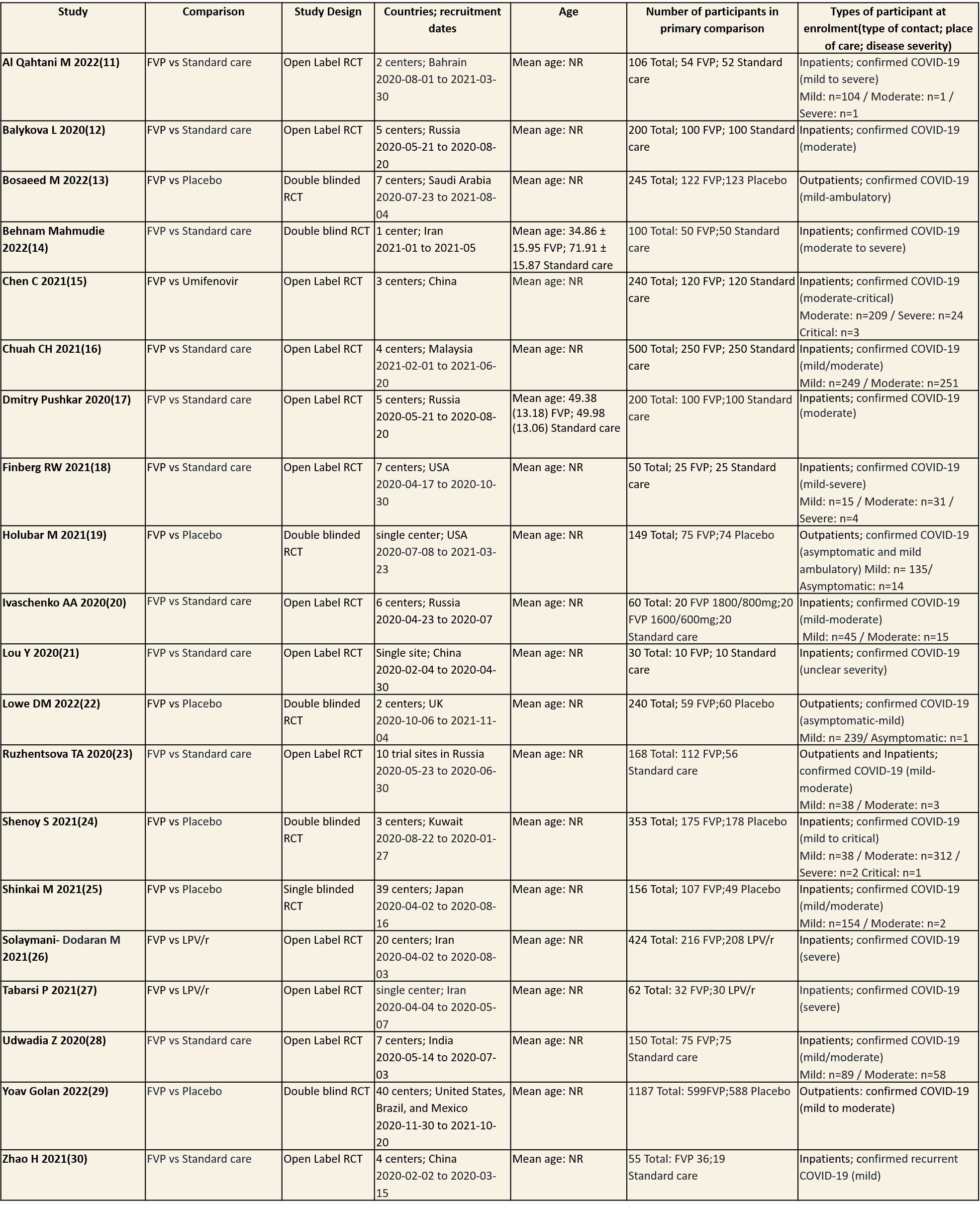
We found 20 RCTs that matched the PICO question as pre-defined by the expert working group. The trials included a total of 4675 participants, all of whom were adults. Three trials were conducted in China, early in the pandemic; all completed recruitment in April 2020(15,21,30). The other trials recruited from April 2020 to November 2021: in Iran (13,26,27); Russia (12,17,20,23); India(28); Bahrain (11); Saudi Arabia(14); Malaysia (16); the UK (22); the USA (18,19); Kuwait (24); Japan(25);one trial recruited from the United states of America, Brazil and Mexico(29) . all trials varied in disease severity ranging from mild to critical. 15 trials recruited hospitalized patients (11-13,15-18,20,21,24-30), 1 trial recruited both outpatients and inpatients(23) whilst the other four trials were focused on ambulatory care and only included outpatients (14,19,22,29).
Severity of COVID‐19 disease at enrolment was reported as asymptomatic, mild, moderate, severe or critical; this was inferred using classification as described by the authors in accordance with the WHO guidance. Of the 4675 participants (20 trials), 15 (0.3%) were asymptomatic, 1412 (30.2%) had mild disease, 1410 (30.1%) moderate disease, 517 (11.1%) severe disease and 4(0.08) had critical disease. Severity was either unclear or not reported in 1317(28.1%) of participants.
Each trial and its results are described below; characteristics of the trials are summarised in the Summary of characteristics of included trials table. All the trials have varying risk of bias across all domains. Risk of bias for each domain per trial is displayed alongside the forest plots below.
The following comparisons were investigated in the trials (we compared outcomes for arms randomized to Favipiravir vs. standard of care or active comparators).
1. Favipiravir (FVP) vs Standard of care without Favipiravir/Placebo
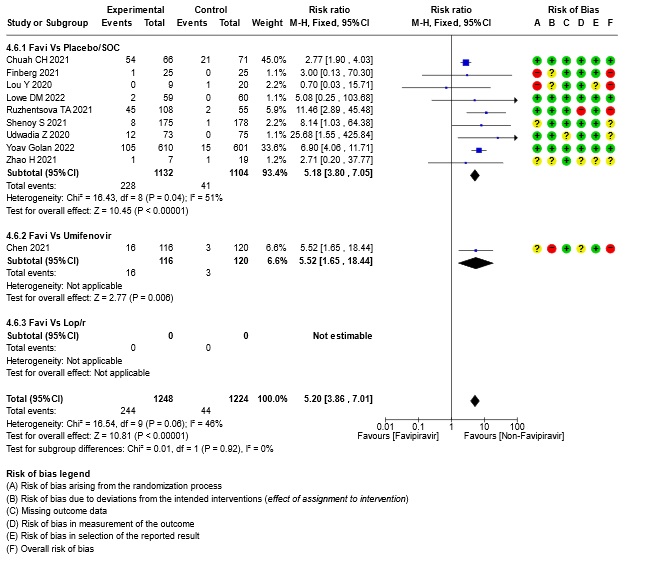
Seventeen trials were included in this comparison (11-13,16-25,28-30). Twelve trials compared FVP to standard of care (11-13,16-21,23,25,28,30) whereas five trials, (14,19,22,24,29), compared FVP to placebo.
2. Favipiravir vs Umifenovir
One Trial was included in this comparison (15).
3. FVP vs Lopinavir/ritonavir
Two trials were included in this comparison, in which participants were randomized 1:1 ratio to receive FVP or LPV/r (26,27)
Since Favipiravir was compared with Standard of care or placebo or an active comparator we did a sensitivity analysis to assess the robustness of the pooled estimate by stratifying the comparators separately from standard of care or placebo. We did this for critical outcomes of mortality, time to clinical improvement, time to negative PCR and duration of hospitalization. We concluded that there was no difference between the pooled estimate and the analysis done separately for Favipiravir vs SOC/placebo and Favipiravir vs Active comparator and hence decided to use the pooled estimate for representing the meta-analysis and summary of findings tables.
Our expert working group classified progression to oxygen, non-invasive ventilation (NIV) and invasive mechanical ventilation (IMV) as critical outcomes and mortality, time to clinical improvement and thrombotic or secondary infections as important outcomes. However, as the situation in the country evolved, the guidelines group upgraded mortality, progression to respiratory failure, oxygen requirement and critical and intensive care admission as critical outcomes and others as important outcomes.
Critical (primary) Outcomes
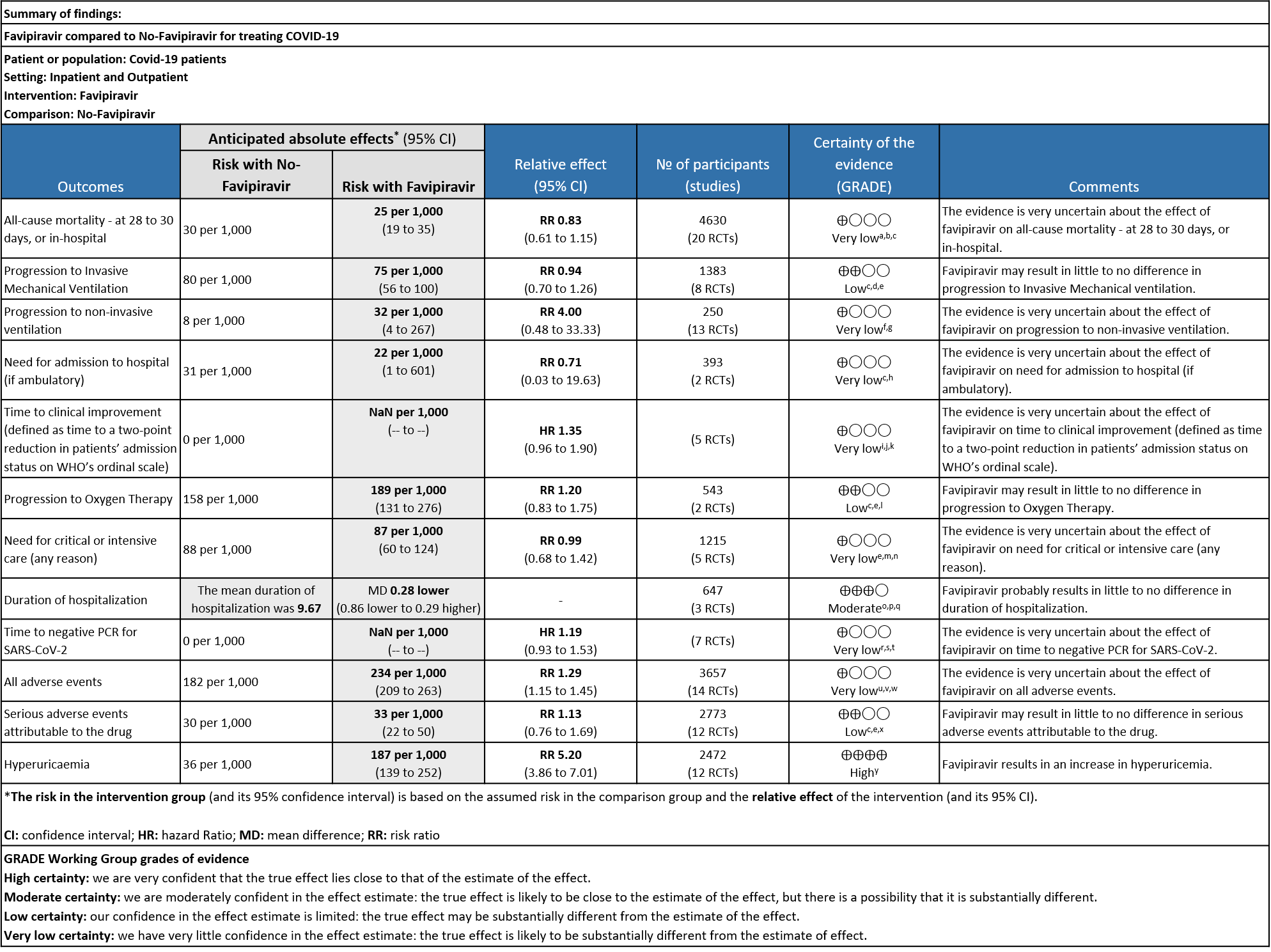
As presented in the ‘Summary of findings’ table, the evidence is of very low certainty for the effect of Favipiravir on mortality, progression to non-invasive ventilation, progression to invasive mechanical ventilation, time to clinical improvement, need for hospital admission, time to negative PCR and adverse events-all and serious. The outcomes of duration of hospitalization were regarded as moderate certainty evidence and adverse event of hyperuricemia as high certainty evidence.
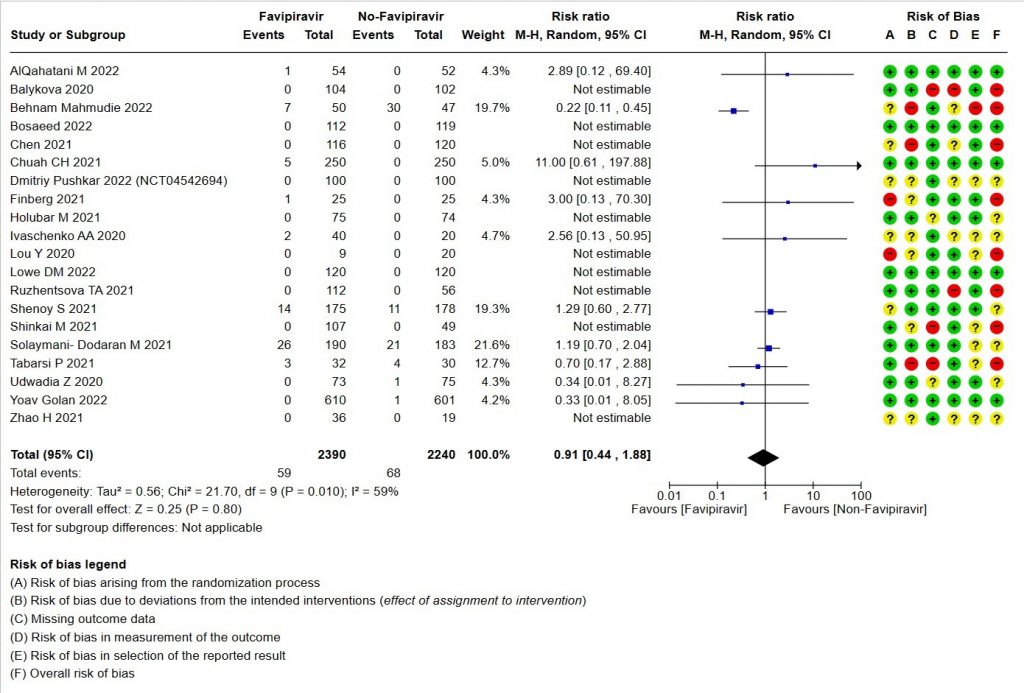
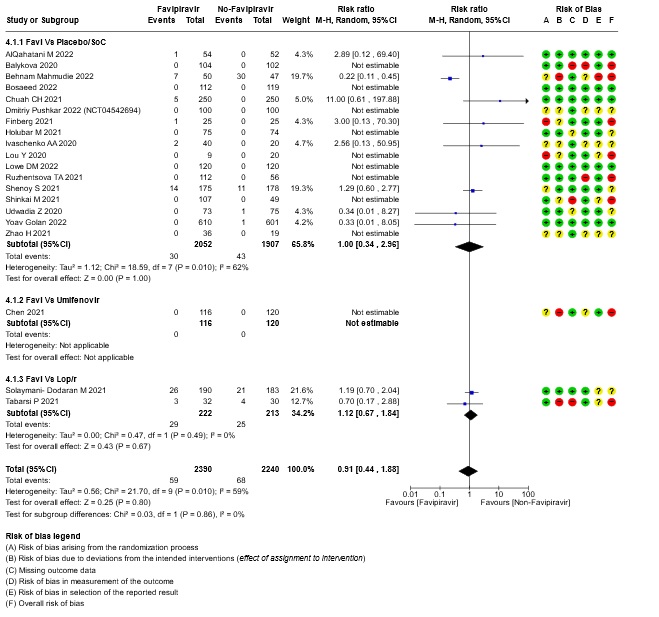
- All-cause mortality: Very low certainty of evidence in 4630 patients in twenty RCTs(11-30) found the evidence is very uncertain about the effect of Favipiravir on all-cause mortality vs. standard of care or active comparator (RR 0.91; 95% CI 0.44 to 1.88). Sensitivity analysis performed with the sole inclusion of low risk of bias studies did not demonstrate an appreciable change in the statistical significance of the pooled estimate results (RR 2.38, 95% CI [0.31, 18.19]).
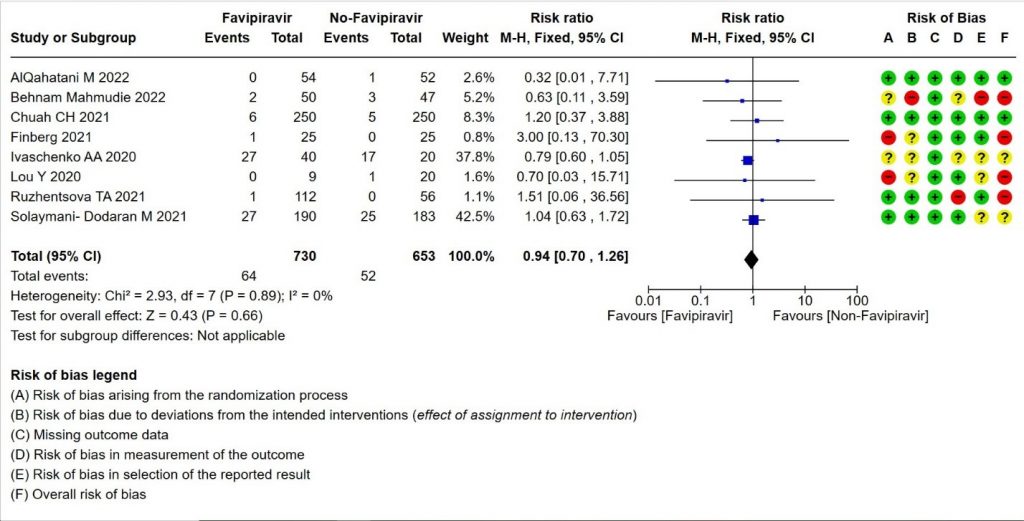
- Progression to Invasive mechanical ventilation:Low certainty evidence in 1383 patients from eight RCTs (11,13,16,18,20,21,23,26) found that the Favipiravir may result in little to no difference in progression to Invasive Mechanical ventilation (RR 0.94; 95% CI 0.70 to 1.26).
- Progression to non-Invasive ventilation:Very low certainty evidence in 250 patients from thirteen RCTs (11-22,29) found that the evidence is very uncertain about the effect of Favipiravir vs. standard of care or active comparator on progression to non-invasive ventilation (RR 4.0; 95% CI 0.48 to 33.33).
- Need for hospital admission: Very low certainty in 393 patients from two RCTs(14,19)found that the evidence is very uncertain for Favipiravir standard of care or active comparator on the need for hospitalization (RR 0.71; 95% CI 0.03 to 19.63).
- Time to clinical improvement (>2 point reduction in the WHO ordinal score): Very low certainty in 921 patients from five RCTs (12,18,23,24,28) found that the evidence is very uncertain about the effect of Favipiravir standard of care or active comparator on time to clinical improvement (HR 1.35; 95% CI 0.96 to 1.90). Sensitivity analysis performed with removal of studies with high risk of bias did not change the pooled estimate effect (RR 1.18 [0.89 , 1.56])
- Progression to oxygen therapy: Low certainty evidence in 543 patients from two studies(16,21), found that Favipiravir may result in little to no difference in progression to Oxygen Therapy when compared to standard of care or active comparator (RR 1.20; 95% CI 0.83 to 1.75).
- Need for critical or intensive care for any reason:Very low certainty evidence in 1215 patients from five RCTs (11,16,23,26,27), found that Favipiravir found that the evidence is very uncertain about the effect of Favipiravir on progression to intensive care when compared to standard of care or active comparator (RR 0.99; 95% CI 0.68 to 1.42).
- Duration of hospitalization: Moderate certainty of evidence in 647 patients from three RCTs (13,16,18) reported that Favipiravir probably results in little to no difference in duration of hospitalization. Pooled duration of hospitalization was similar in both groups i.e. it did not differ between participants who received FVP and those who did not (mean difference (MD) 0.28 days shorter with FVP, 95% CI 0.86 lower to 0.29 higher.
- Time to negative PCR: Very low certainty of evidence from seven RCTs(14,18,19,23,25,28,30) found that the evidence is very uncertain for effect of Favipiravir on time to negative PCR when compared to standard of care or active comparator (HR 1.19 95% CI 0.93 to 1.53). Sensitivity analysis performed after removing studies with high risk of bias (18,23,25) had minimal effect on the pooled hazard ratio (HR: 1.13 [0.76 , 1.68] 4 RCTs).
- All Adverse events : Very low certainty of evidence in 3657 patients from fourteen RCTs(12,14-16,18-20,22-25,28,20) found that the evidence is very uncertain on effect of favipiravir on adverse events when compared to standard of care or active comparator (RR 1.29; 95%CI 1.15 to 1.45). sensitivity analysis which incorporated removal of studies with high risk of bias (12,15,18,23,25) demonstrated pooled effect estimate that was not statistically significant (RR 1.40 [1.00 , 1.97]). Sensitivity analysis incorporating a random effects model also did not change the pooled estimate effect significantly (RR 1.32 [1.04 to 1.66] 14RCTs).
- Serious Adverse events (attributable to the drug): Very low certainty of evidence in 2773 patients from twelve RCTs(12,14,15,19,21,22-25,28,30) found that Favipiravir may result in little to no difference in serious adverse events attributable to the drug when compared to standard of care or active comparator (RR 1.13; 95%CI 0.76 to 1.69).
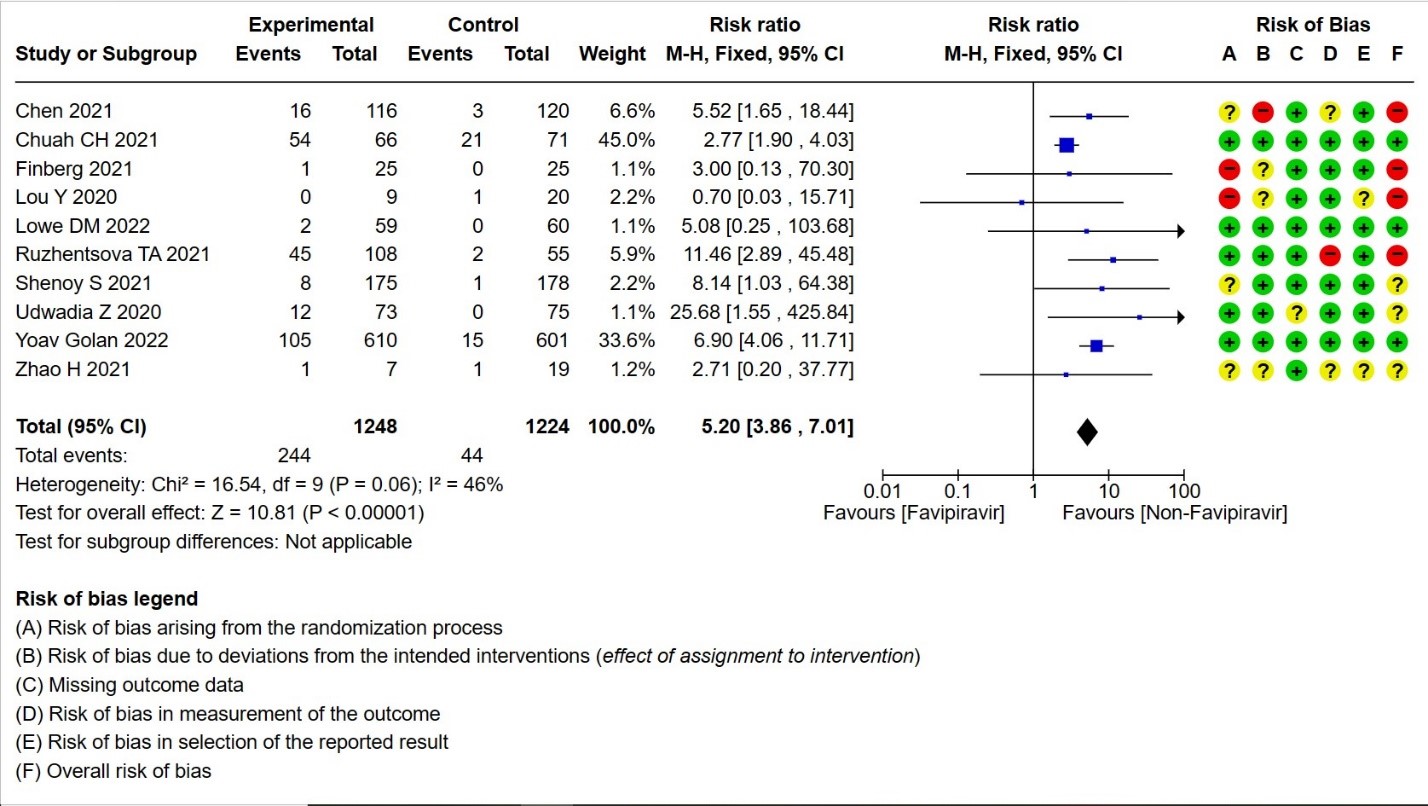
- Adverse Events – Hyperuricemia: High certainty of evidence in 2472 patients from twelve RCTs (15,16,18,21-24,28-30) found that Favipiravir results in an increase in hyperuricemia (RR 5.20; 95%CI 3.86 to 7.01).
1. All‐cause mortality ‐ at 28 to 30 days, or in hospital.
2. Progression to Invasive Mechanical Ventilation
3. Progression to non‐invasive ventilation
4. Need for admission to hospital (if ambulatory)
5. Time to clinical improvement (defined as time to a two‐point reduction in patients’ admission status on WHO’s ordinal scale).
6. Progression to Oxygen Therapy
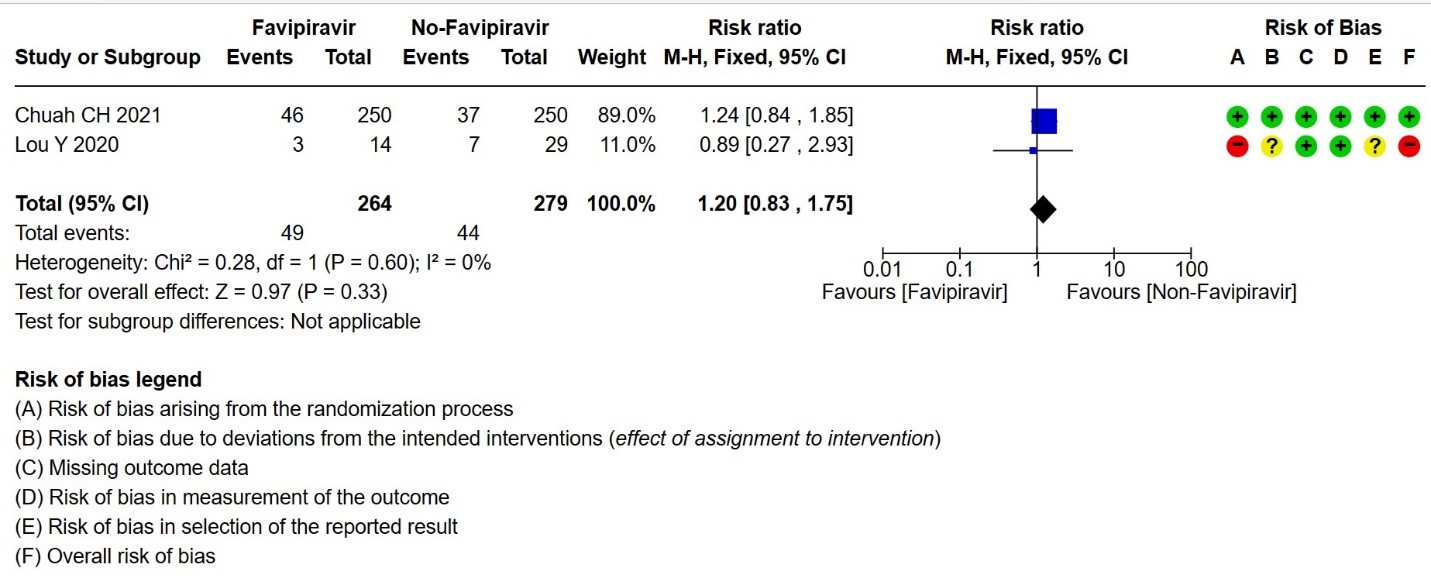
7. Need for critical or intensive care (any reason)
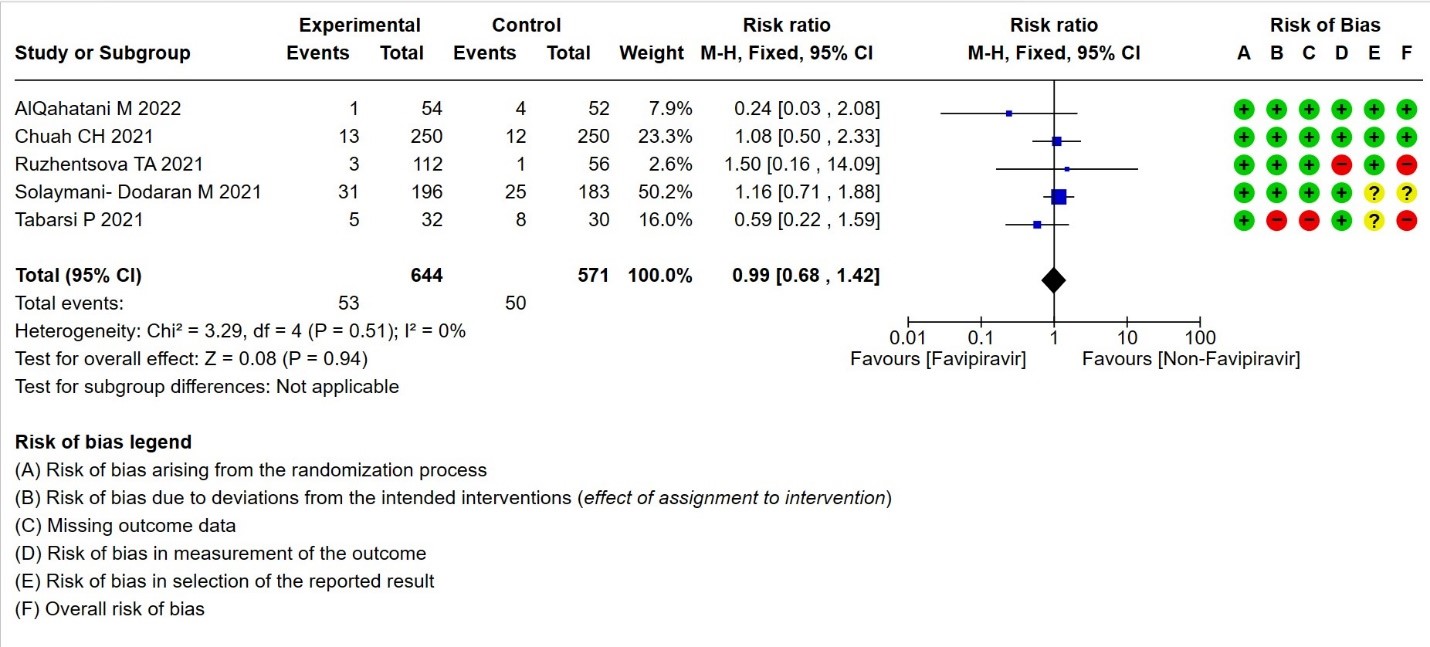
8. Duration of hospitalization
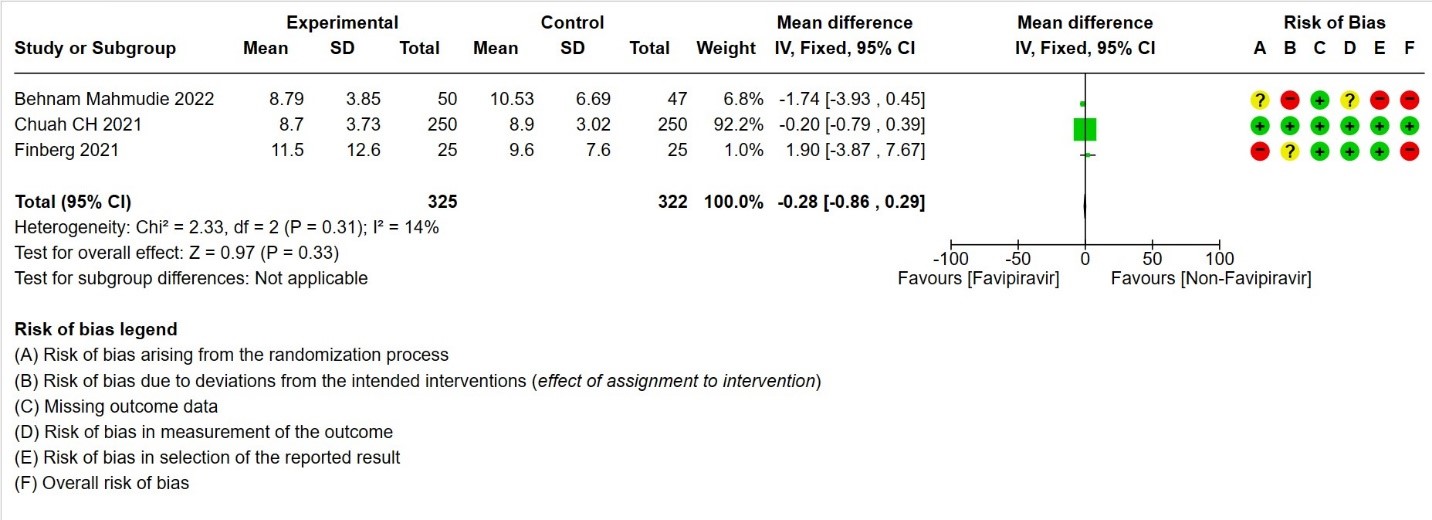

9. Time to negative PCR for SARS‐CoV‐2
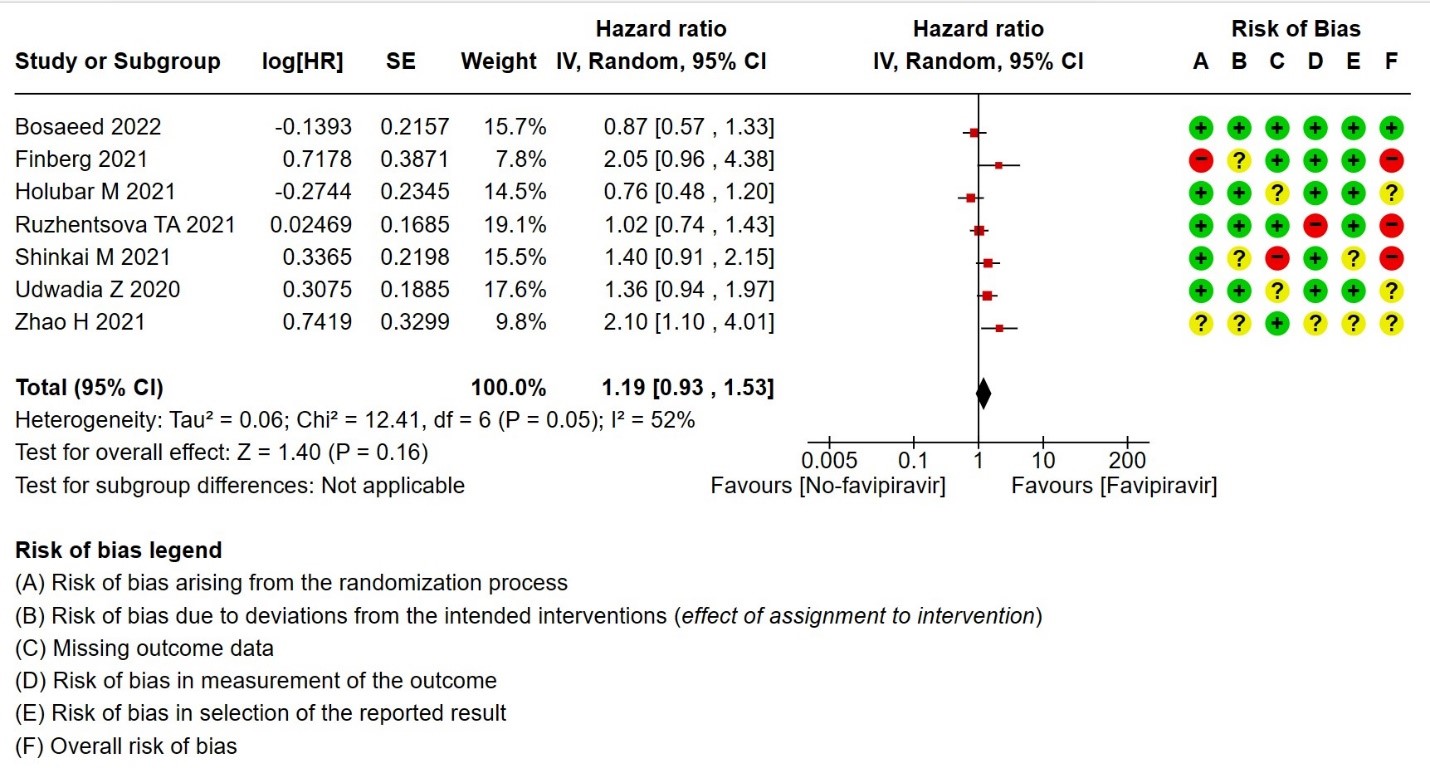
10. All adverse events
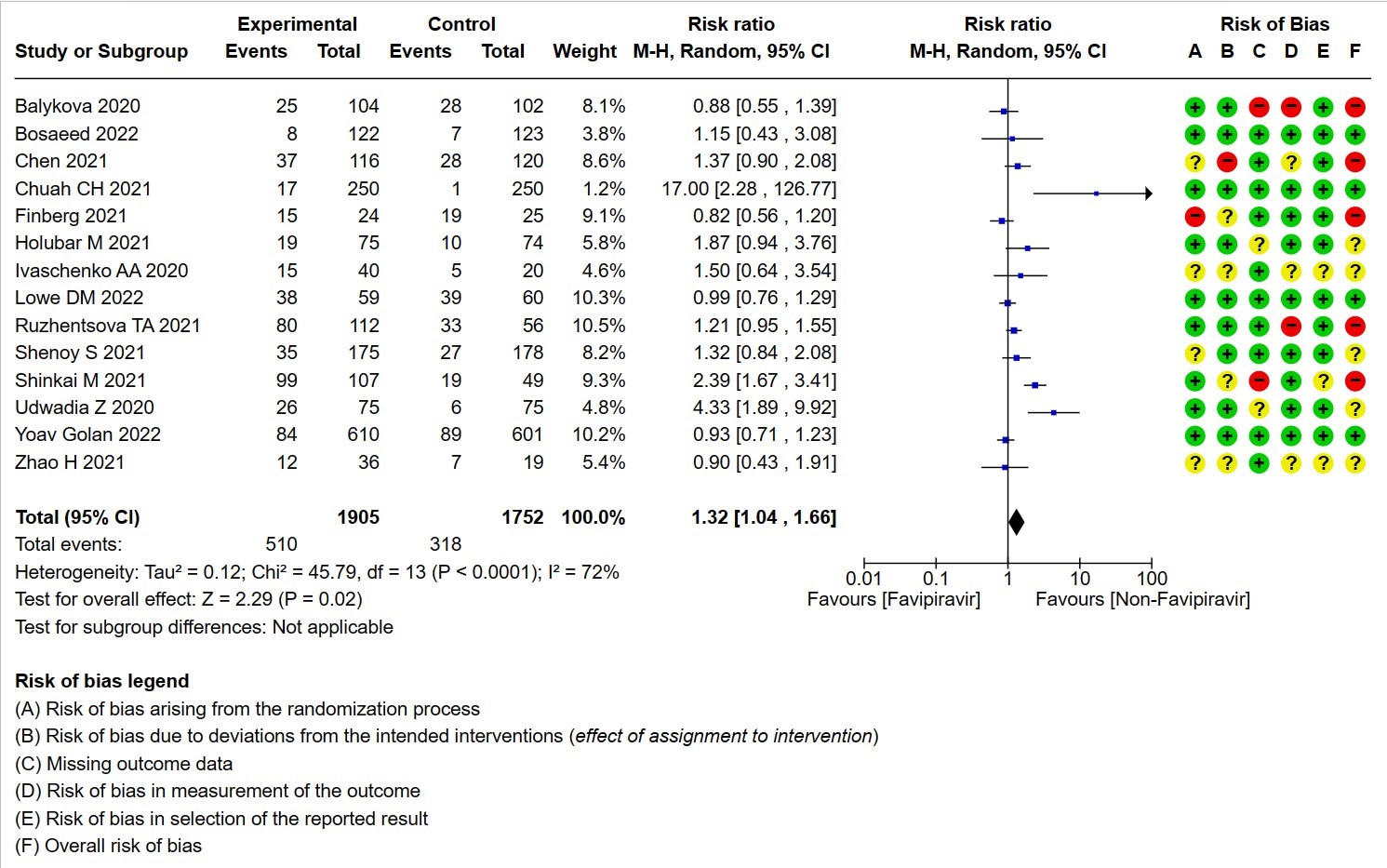
11. Serious adverse events attributable to the drug
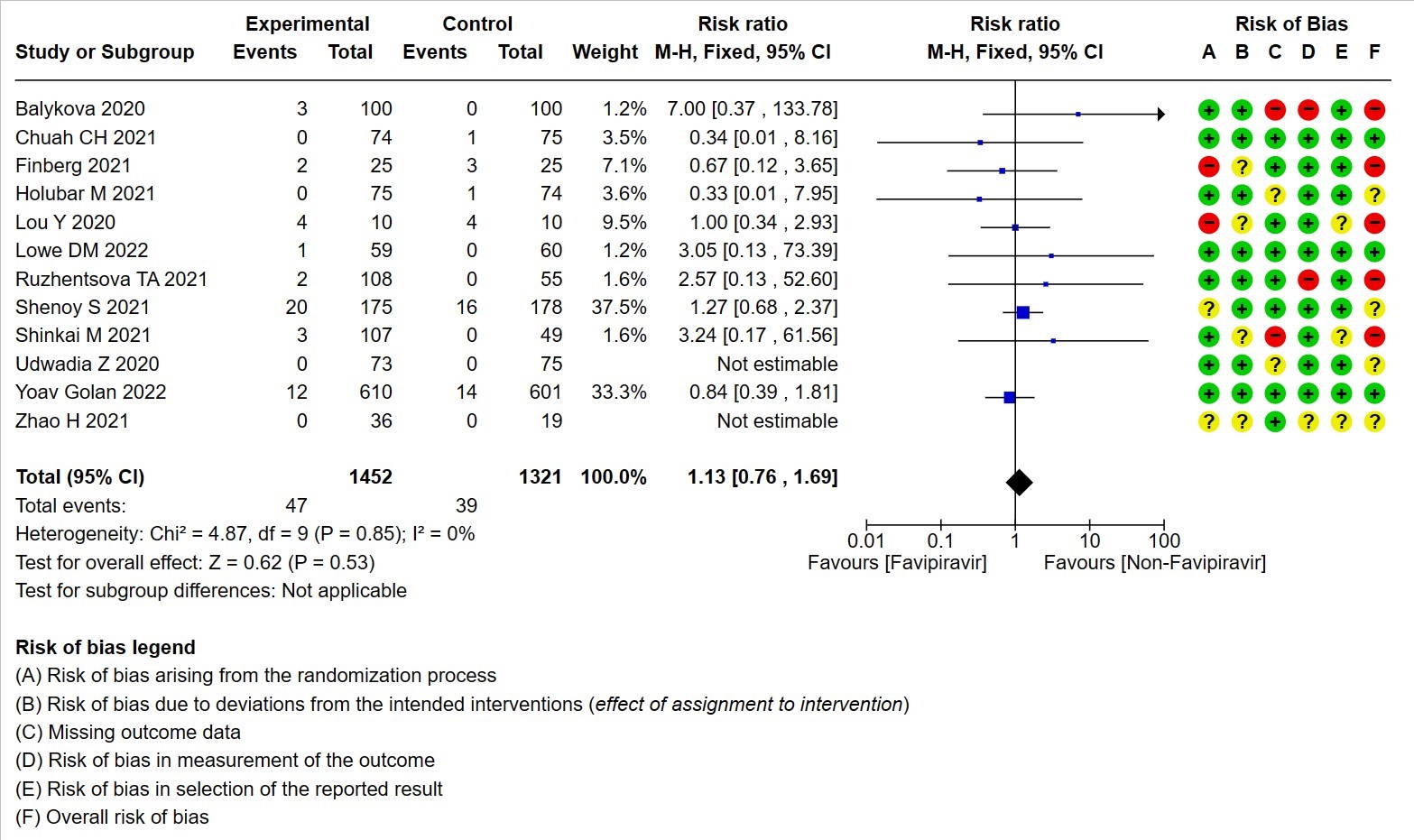
12. Hyperuricemia
Sensitivity analysis (SOC + active comparator (another antiviral) separately vs together)
13. Subgroup analysis: - All-cause Mortality
14. Subgroup analysis: - Need for Critical Care
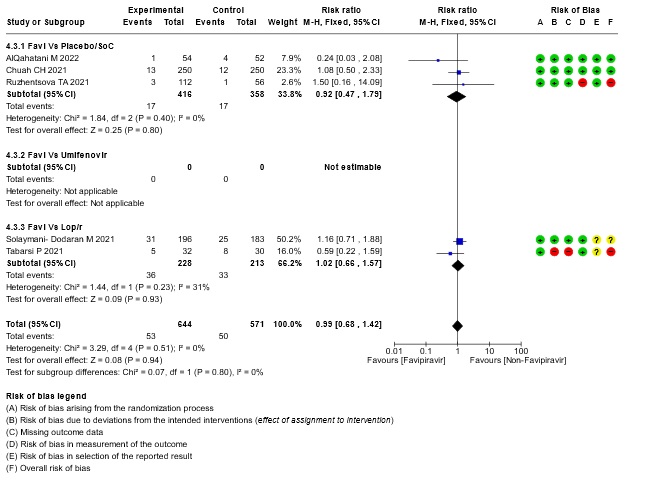
15. Subgroup analysis :- Time to negative PCR
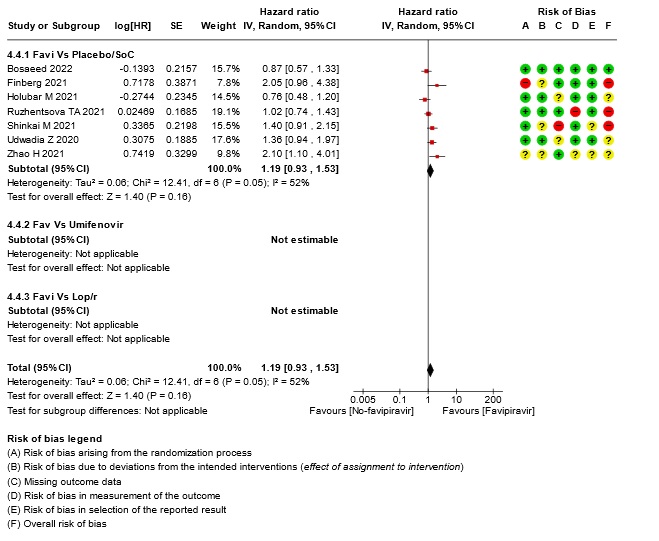
16. Subgroup analysis: - Time to Clinical Improvement
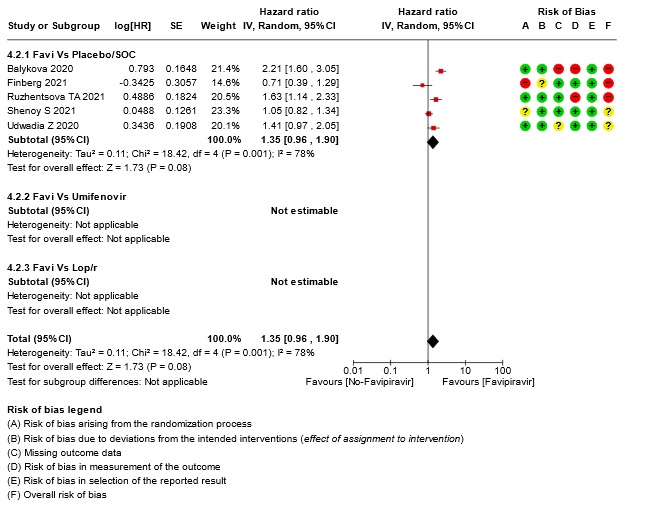
17. Subgroup analysis: - All-Adverse Events
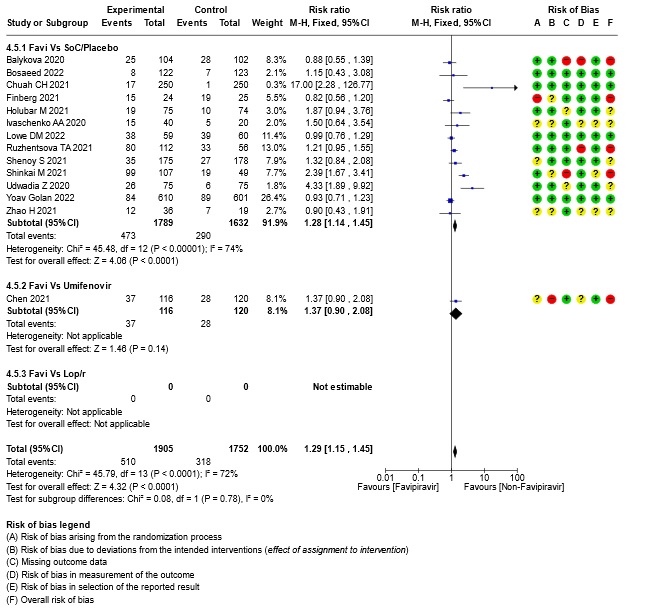
18. Subgroup analysis: - Hyperuricemia